百匯澤®是一款強效、高選擇性的 PARP1 和 PARP2 抑制劑,在晚期 HER2 陰性乳腺癌患者中顯示出有意義且持久的有效性
百匯澤®對比安慰劑在胃癌患者中顯示出數值上更優的無進展生存期,但未達到統計學顯著性差異
在兩項試驗中,百匯澤®總體耐受性均良好
百匯澤®近日在中國獲批用於治療既往接受過治療的晚期卵巢癌患者
中國北京和美國麻省劍橋2021年6月4日 /美通社/ -- 百濟神州(納斯達克代碼:BGNE;香港聯交所代碼:06160)是一家全球化的生物科技公司,專注於在世界範圍內開發和商業化創新藥物。公司今日宣佈,其PARP抑制劑百匯澤®(帕米帕利)顯示出治療HER2陰性乳腺癌患者的有效性,在胃癌中顯示出數值上更優的無進展生存期,但未達到統計學顯著性差異,此項結果可能與胃癌相關試驗未能達到計劃的入組目標有關。這些數據將在2021年6月4日至8日舉行的美國臨床腫瘤學會年會(ASCO 2021)進行海報展示,包括一項評估百匯澤®治療胚系BRCA1/2突變(gBRCA1/2m)的局部晚期或轉移性HER2陰性(HER2[-])乳腺癌患者的2期試驗的初期報告,以及百匯澤®作為對鉑類藥物一線化療有緩解的不可手術的局部晚期或轉移性胃癌患者維持治療的隨機2期試驗的初期報告。
百濟神州腫瘤免疫學首席醫學官賁勇醫學博士表示:「我們很高興能分享百匯澤®臨床開發項目的進展,尤其在那些高發的疾病領域,這與百濟神州致力於改善患者生存現狀的願景一脈相承。百匯澤®在HER2陰性乳腺癌中的陽性結果令人振奮,HER2陰性乳腺癌是一種致命的惡性腫瘤,也是女性癌症死亡的主要原因。儘管百匯澤®在胃癌中的試驗結果未達到統計學顯著性差異,但在推進百匯澤®的全球開發過程中,我們仍希望這些結果能夠推動科學界的認知,併為百匯澤®的耐受性提供了進一步的依據。」
百匯澤®治療伴gBRCAm的局部晚期或轉移性的HER2陰性乳腺癌患者的2期試驗結果
海報編號1087
本項單臂、開放性、多中心2期試驗(NCT03575065)旨在評價百匯澤®在攜帶有害或疑似有害gBRCA1/2m,且既往接受過不超過兩線化療的局部晚期或轉移性HER2陰性乳腺癌患者中的安全性和有效性。試驗共入組88例患者,包括62例三陰性乳腺癌患者(TNBC佇列)和26例激素受體陽性(HR[+])和HER2(-)乳腺癌患者(HR[+]佇列)。根據獨立審查委員會(IRC)評估,TNBC佇列的55例患者和HR(+)佇列的21例患者在基線時具有可測量病灶。試驗的主要終點為IRC根據RECIST 1.1版評估的客觀緩解率(ORR);次要終點包括研究者評估的ORR、緩解持續時間(DoR)、最佳總體緩解(BOR)、無進展生存期(PFS)、IRC和研究者評估的臨床獲益率(CBR)和疾病控制率(DCR),以及總生存期(OS)、安全性和耐受性。
中國醫學科學院腫瘤醫院醫學博士兼該項試驗的主要研究者徐兵河教授表示:「研究表明,攜帶胚系BRCA突變的乳腺癌可能對PARP抑制劑敏感。 2期臨床結果顯示百匯澤®在HR(+)/HER2(-)及三陰性乳腺癌患者中展示出臨床療效,而三陰性乳腺癌被稱為是最具侵襲性且預後最差的乳腺癌。臨床結果進一步表明,百匯澤®不僅能帶來頗高的緩解率,還有望帶來無進展生存期獲益,我們期待能在這些亟需更多治療方案的人群中展開進一步的研究。」
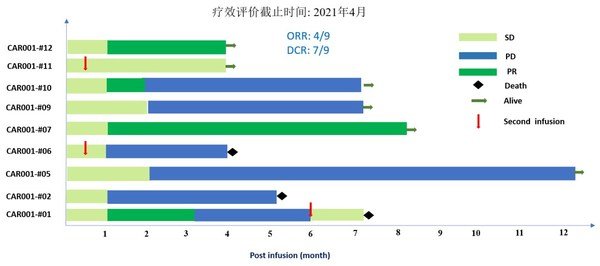
截至數據截止日期2020年10月9日,中位隨訪時間為13.8個月(TNBC佇列,10.9個月;HR[+]佇列,18.5個月)。
百匯澤®在兩個佇列的患者中均表現出有臨床意義且持久的活性。有效性結果包括:
- IRC評估的經確認的ORR(主要有效性終點)在HR(+)隊列為61.9%(95% CI:38.4,81.9),TNBC隊列為38.2%(95% CI:25.4,52.3)
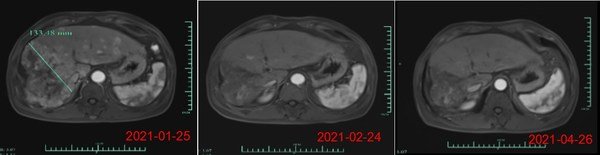
- 共4例患者達到完全緩解(CR),包括TNBC佇列3例患者和HR(+)佇列1例患者
- TNBC佇列18例患者和HR(+)佇列12例患者達到部分緩解(PR)
- IRC評估的DCR在HR(+)隊列為90.5%(95% CI:69.6,98.8),TNBC隊列為72.7%(95% CI:59.0,83.9)
- 此外,IRC評估的CBR在HR(+)隊列為71.4%(95% CI:47.8,88.7),TNBC隊列為43.6%(95% CI:30.3,57.7)
- 中位DoR在HR(+)隊列為7. 5個月(95% CI:5.6,14.8),TNBC隊列為7.0個月(95% CI:3.9,不可估計[NE])
- 中位PFS在HR(+)隊列為9.2個月(95% CI:7.4,11.9),TNBC隊列為5.5個月(95% CI:3.7,7.3)
- TNBC佇列的中位OS為17.1個月(95% CI:13.7,NE),HR(+)佇列未達到中位OS(NE;95% CI:18.1,NE)
百匯澤®總體耐受性良好,兩個佇列所有88例患者的安全性分析結果為:
- 87例患者(98.9%)出現至少1起任何級別的治療中出現的不良事件(TEAE),54例患者(61.4%)出現至少1起3級及以上的TEAE
- 87例患者(98.9%)出現至少1起任何級別的治療相關TEAE,53例患者(60.2%)出現至少1起3級及以上的治療相關TEAE,最常見的TEAE(≥5%)為貧血(3 9.8%)、中性粒細胞計數降低(29.5%)、白細胞計數降低(21.6%)、血小板計數降低(9.1%)、白細胞減少症(5.7%)和中性粒細胞減少症(5.7%)
- 19例患者(21. 6%)出現嚴重TEAE,15例患者(17.0%)出現嚴重治療相關TEAE
- 2例患者因TEAE而終止治療,均與治療相關
- 1例患者(1.1%)因TEAE死亡,未出現導致死亡的治療相關TEAE
百匯澤®對比安慰劑治療局部晚期或轉移性胃癌的2期試驗結果
海報編號3109
PARALLEL 303是一項雙盲、隨機、多中心2期試驗(NCT03427814),百匯澤®對比安慰劑作為維持療法用於治療對鉑類藥物一線化療有緩解的不可手術的局部晚期或轉移性胃癌患者的安全性和有效性。試驗共入組136例患者,由於入組耗時長,且該患者人群的標準治療發生變化,本試驗未達到計劃的約540例患者入組目標。患者以1:1的比例隨機到兩組,接受百匯澤®(n=71)或安慰劑(n=65)60 mg口服,每日兩次,每28天為一個週期。主要終點為研究者根據RECIST 1.1版評估的PFS;次要終點包括至後續治療的時間(TSST)、研究者評估的ORR、DoR和至緩解時間(TTR)、OS和安全性。在進行數據分析時,OS數據尚不成熟。
義大利那不勒斯第二大學Fortunato Ciardiello醫學博士兼試驗主要研究者表示:「許多胃癌患者在疾病後期對目前可用的療法產生了耐藥性,持續進行研究對於尋找有望改善患者預后和生存的藥物至關重要。儘管PARALLEL 303研究未顯示出統計學顯著性差異,但試驗結果有助於我們深入瞭解PARP抑制劑在轉移性胃癌中的作用,進一步支持了百匯澤®的安全性特徵和針對合適患者人群的潛在臨床獲益。」
截至數據截止日期2020年12月7日,中位隨訪時間為8.0個月(百匯澤®組7.9個月;安慰劑組8.0個月)。
百匯澤®組的中位PFS為3.7個月(95% CI:1.9,5.3),高於安慰劑組的2.1個月(95% CI:1.9,3.8),但差異未達到統計學顯著性(p=0.1428;HR=0.799[95% CI:0.5,1.2])。其他有效性結果包括:
- 百匯澤®組的中位OS為10.2個月(95% CI:8.7,16.3),安慰劑組為12.0個月(95% CI:8.2,無法估計[NE])
- 百匯澤®組的ORR為7.7%(95% CI:1.6,20.9),安慰劑組為6.3%(95% CI:0.8,20.8)
- 百匯澤®組的中位DoR為3.6個月(95% CI:3.5,NE),安慰劑組為NE(95% CI:5.6,NE)
- 百匯澤®組的中位TTR為3.7個月(範圍:1.8,7.3),安慰劑組為1.9個月(範圍:1.9,1.9)
在本試驗中,百匯澤®總體耐受性良好,未觀察到新的安全性警示。安全性結果包括:
- 百匯澤®組有65例患者(91.5%)出現至少1起任何級別的TEAE,安慰劑組有61例患者(93.8%)
- 百匯澤®組分別有14例(19.7%)和29例(40.8%)患者出現嚴重TEAE和3級及以上TEAE,安慰劑組分別有10例(15.4%)和20例患者(30.8%)
- 百匯澤®組中最常見(≥10%)的TEAE包括貧血(36.6%)、噁心(32.4%)、食慾下降(26.8%)、嘔吐(23.9%)、乏力(21.1%)、腹瀉(18.3%)、上腹痛(16.9%)、天門冬氨酸氨基轉移酶升高(AST;12.7%)、丙氨酸氨基轉移酶升高(ALT;11.3%)、腹痛(11.3%)、便秘(11.3%)和白細胞計數降低(11.3%)
- 安慰劑組中最常見(≥ 10%)的TEAE包括腹痛(18.5%)、噁心(16.9%)、外周感覺神經病(13.8%)、乏力(16.9%)、貧血(12.3%)、食慾下降(12.3%)、吞咽困難(12.3%)、上腹痛(10.8%)、便秘(10.8%)和腹瀉(10.8%)
- 百匯澤®組有8例患者(11.3%)因TEAE而終止治療,安慰劑組有2例(3.1%)
- 百匯澤®組有2例患者(2.8%)因TEAE死亡,均認定與治療無關;安慰劑組有2例患者(3.1%)(其中1例[1.5%]的TEAE認定與治療相關)
欲瞭解更多關於百濟神州研發和ASCO會議活動的信息,請訪問 https://beigenevirtualexperience.com/。
關於百匯澤®
百匯澤®(帕米帕利)是一款PARP1和PARP2抑制劑,臨床前模型顯示其具有穿透血腦屏障和捕獲PARP-DNA複合物等藥理學特性。由百濟神州的科學家在北京研發中心自主研發,百匯澤®目前正作為單一療法或與其他藥物聯用治療多種惡性實體瘤進行全球臨床開發。迄今為止,已有1200多例患者入組百匯澤 ®臨床試驗。
2021年5月,中國國家藥品監督管理局(NMPA)附條件批准百匯澤®用於治療既往接受過至少兩線化療、攜有胚系BRCA(gBRCA)突變的晚期卵巢癌、輸卵管癌或原發性腹膜癌患者。針對該適應症的完全批准將取決於正在開展中的確證性臨床試驗結果。
百匯澤®臨床項目
百匯澤®的臨床試驗包括:
- 在中國開展的帕米帕利對比安慰劑用於鉑敏感的復發性卵巢癌患者維持治療的3期臨床試驗(NCT03519230)
- 帕米帕利用於治療攜有同源重組缺陷轉移性去勢抵抗性前列腺癌患者的2期臨床試驗(NCT03712930)
- 在中國開展的帕米帕利用於治療攜有BRCA突變的轉移性HER2陰性乳腺癌患者的2期臨床試驗(NCT03575065)
- 帕米帕利用於治療晚期或不可手術的胃癌患者的2期臨床試驗(NCT03427814)
- 在中國開展的帕米帕利用於治療晚期卵巢癌、輸卵管癌、原發性腹膜癌或晚期三陰乳腺癌患者的1/2期臨床試驗(NCT03333915)
- 帕米帕利聯合放療及/或替莫唑胺用於治療新診斷或復發/難治性多形性膠質母細胞瘤患者的1b/2期臨床試驗(NCT03150862)
- 帕米帕利聯合替莫唑胺用於治療局部晚期或轉移性實體瘤患者的1b期臨床試驗(NCT03150810)
- 帕米帕利聯合百澤安用於治療多項惡性實體瘤的1b期臨床試驗(NCT02660034)
關於百濟神州腫瘤
百濟神州通過自主研發或與志同道合的合作夥伴攜手,不斷推動同類最佳或同類第一的臨床候選藥物研發,致力於為全球患者提供有影響力、可及且可負擔的藥物。公司全球臨床研究和開發團隊已有約2300人,團隊規模還在不斷擴大。這支團隊目前正在全球範圍支持開展90多項臨床研究,已招募患者和健康受試者超過13000人。百濟神州自有的臨床開發團隊規劃並主導公司產品管線的研發和擴充,為覆蓋全球40多個國家/地區的臨床試驗提供支持和指導。公司特別關注血液腫瘤和實體腫瘤的靶向治療及腫瘤免疫治療,並重點研究單葯和聯合療法。目前,百濟神州自主研發的三款藥物已獲批上市:百悅澤®(BTK抑制劑,已在美國、中國、加拿大及其他國際市場獲批上市)、百澤安®(可有效避免Fc-γ受體結合的抗PD-1抗體,已在中國獲批上市)及百匯澤®(已在中國獲批上市)。
同時,百濟神州還與其他創新公司合作,共同攜手推進創新療法的研發,以滿足全球健康需求。在中國,百濟神州正在銷售多款由安進和百時美施貴寶授權的腫瘤藥物。公司也通過與包括安進、百奧泰、EUSA Pharma、Mirati Therapeutics、Seagen以及Zymeworks在內的多家公司合作,更大程度滿足當前全球範圍尚未被滿足的醫療需求。百濟神州還與諾華公司(Novartis Pharma AG)達成合作,授權諾華在北美、歐洲和日本開發、生產和商業化百澤安®。
關於百濟神州
百濟神州是一家立足科學的全球生物科技公司,專注於開發創新、可負擔的藥物,以為全球患者改善治療效果和提高藥物可及性。公司廣泛的藥物組合目前包括40 多款臨床候選藥物,通過強化公司自主競爭力以及與其他公司開展合作,我們致力於加速現有多元、創新藥物管線的開發進程,希望能在2030 年之前為全球20多億人全面改善藥物可及性。百濟神州在全球五大洲打造了一支近6000人的團隊。欲瞭解更多信息,請訪問www.beigene.com.cn。
前瞻性聲明
本新聞稿包含1995年《私人證券訴訟改革法案》Private Securities Litigation Reform Act of 1995和其他聯邦證券法所定義的前瞻性聲明,包括該新聞稿中提及的百匯澤®臨床試驗數據,百匯澤為患者帶來臨床獲益以及在 安全性和耐受性上的優勢的潛能,百濟神州對百匯澤®預期的臨床開發、藥政里程碑和商業化進程,以及在「關於百濟神州腫瘤學」和「關於百濟神州」副標題下提及的百濟神州計劃、承諾、抱負和目標。這些因素包括了以下事項的風險:百濟神州證明其候選藥物功效和安全性的能力;候選藥物的臨床結果可能不支持進一步開發或上市審批;藥政部門的行動可能會影響到臨床試驗的啟動、時程表和進展以及藥物上市審批;百濟神州的上市藥物及候選藥物(如能獲批)獲得商業成功的能力;百濟神州獲得和維護對其藥物和技術的智慧財產權保護的能力;百濟神州依賴第三方進行藥物開發、生產和其他服務的情況;百濟神州取得監管審批和商業化醫藥產品的有限經驗,及其獲得進一步的營運資金以完成候選藥物開發和實現並保持盈利的能力;新冠肺炎全球大流行對百濟神州的臨床開發、監管、商業化運營以及其他業務帶來的影響;以及百濟神州在最近季度報告的10-Q表格中「風險因素」章節里更全面討論的各類風險 ;以及百濟神州向美國證券交易委員會期后呈報中關於潛在風險、不確定性以及其他重要因素的討論。本新聞稿中的所有信息僅及於新聞稿發佈之日,除非法律要求,百濟神州並無責任更新該些信息。